Computational Proteomics Group
Computational Proteomics Group
Computational Proteomics Group
Computational Proteomics Group


Assoc. Prof. Veit Schwämmle
The Computational Proteomics Group develops and applies computational solutions for improved data analysis in large-scale omics experiments with focus on proteins and their post-translational modifications (PTMs). The aim is to better understand the functional protein states in order to determine, confirm and predict their contribution to cell behavior and disease. Our main research interests include
For the full publication record, see Google Scholar.
All our software is open source and available on our repositories. We mostly develop:
R and Python scripts and libraries for functional analysis of omics data
New algorithms for computational mass spectrometry
Deep learning models for MS data
Portable and scalable workflows for HPC and cloud
Statistical methods for proteoform characterizations
The ELIXIR Tools and
Data Service Registry, bio.tools, launched in
January 2015, hosts details about tens of thousands
of software in the life sciences.
Collaboration with the PRIDE repository to enhance metadata annotations using SDRF
We develop web applications for interactive data analysis (see e.g. Web Applications). We have a strong focus on using smart and extensive visualization for deep exploration of omics data. Most of these applications are also available as stand-alone versions via docker and conda, and several as libraries for direct integration into data analysis workflows.











General literature
For an overview
of methods used in proteomics, see our PTMomics review
as well as our editorial about
reproducibility. We also published a paper about transparent reporting of the
different steps in the data analysis workflow: ref.
We published tutorials for
CrossTalkMapper,
VSClust
+ complex analysis and general
PTM analysis, and have further training material on
GitHub.
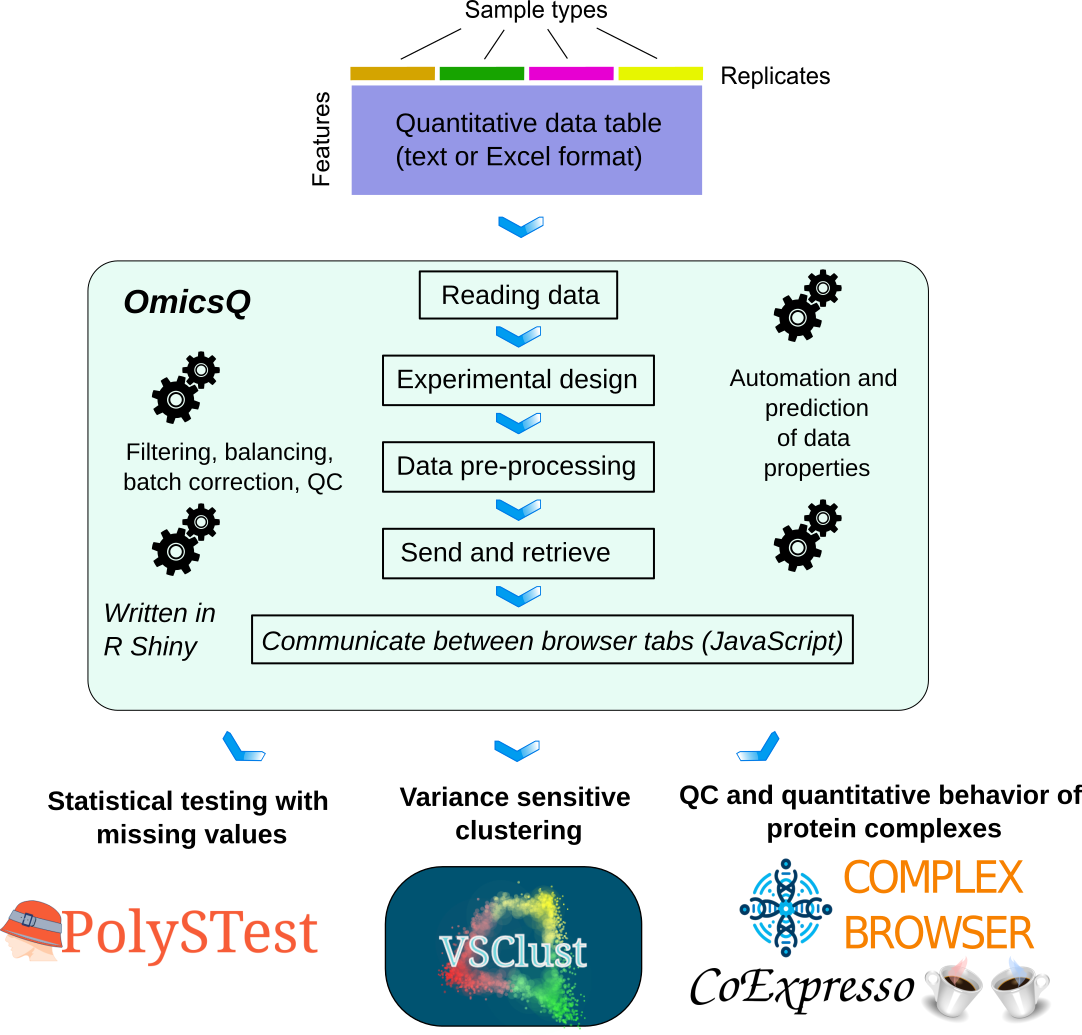
Tool suites for quantitative Omics
Check out our tool suite
OmicsQ facilitating data processing and further analysis with PolySTest,
VSClust et al. (ref).
We also collaborated with the development of a web application for LC-MS metabolomics
data:
MetaboLink.
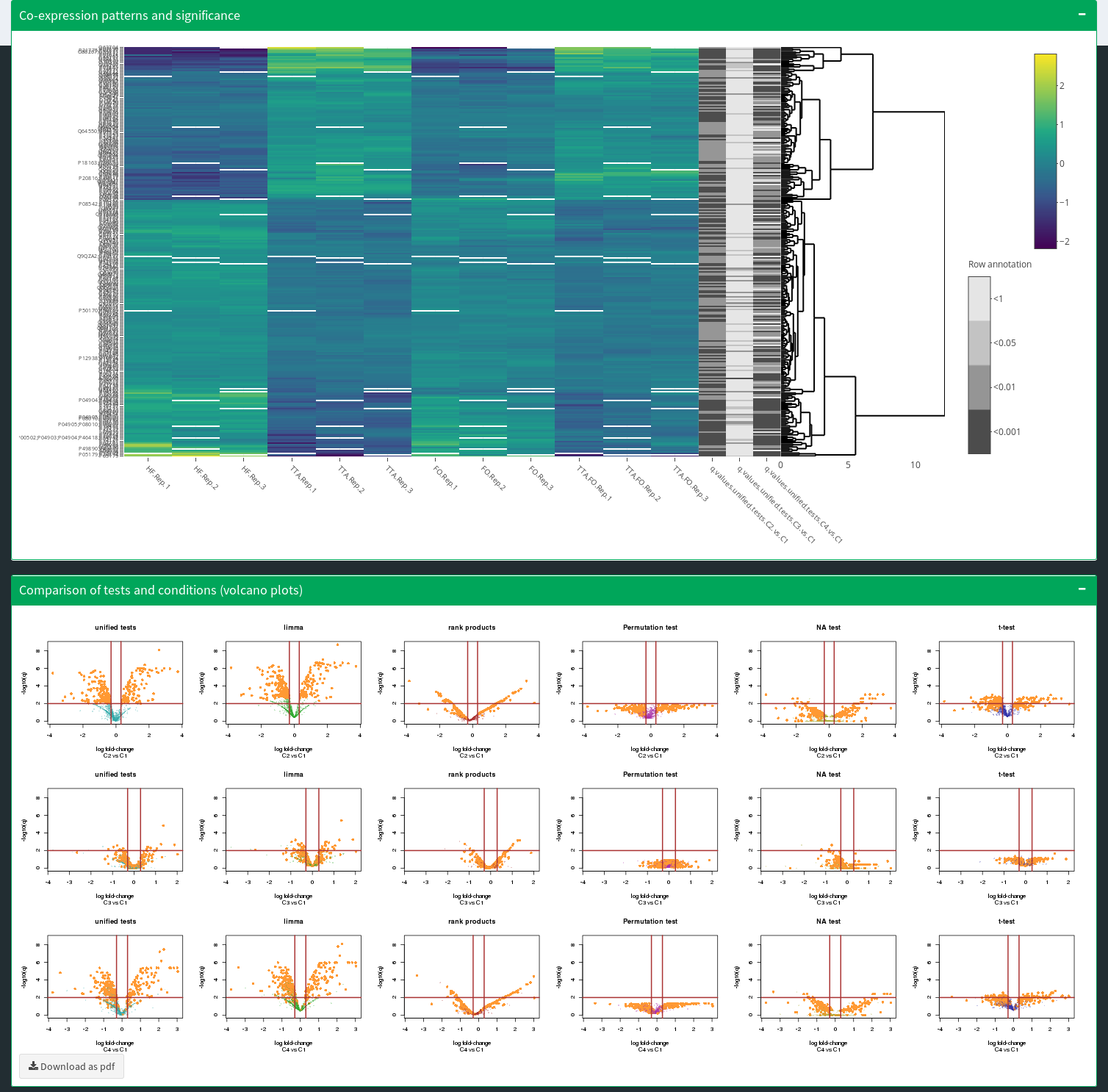
Statistical tests: PolySTest
New statistical test for data with low replicates numbers and many
missing values, combined with
well-performing statistical tests for high
confidence detection of differentially
regulated features (ref.)
PolySTest.
The old LimmaRP approach where we showed the power of combining limma and rank products
(ref.)
is still accessible here:
LimmaRP
Biopharma LC-MS applications
Recent work demonstrates and compares LC-MS-based host-cell-protein quantitation methods
in the USP 1132.1 context, including qualification and benchmarking across workflows
(ref).
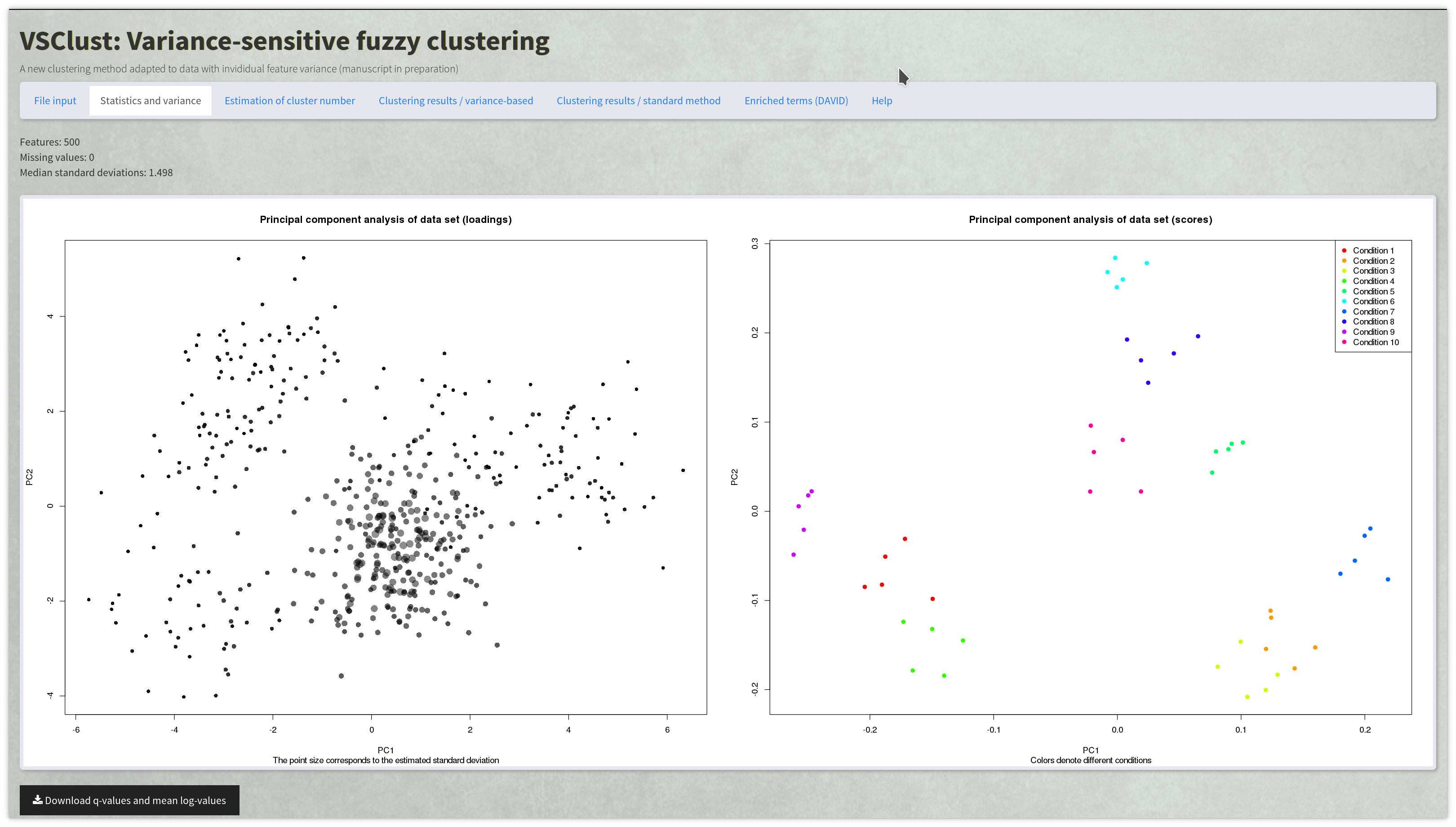
Data clustering
VSClust considers feature variance
leading to more accurate clustering results
(ref.
and tutorial).
VSClust.
See also how to estimate appropriate parameter
values for fuzzy c-means clustering (ref).
(old app:
FuzzyClust)
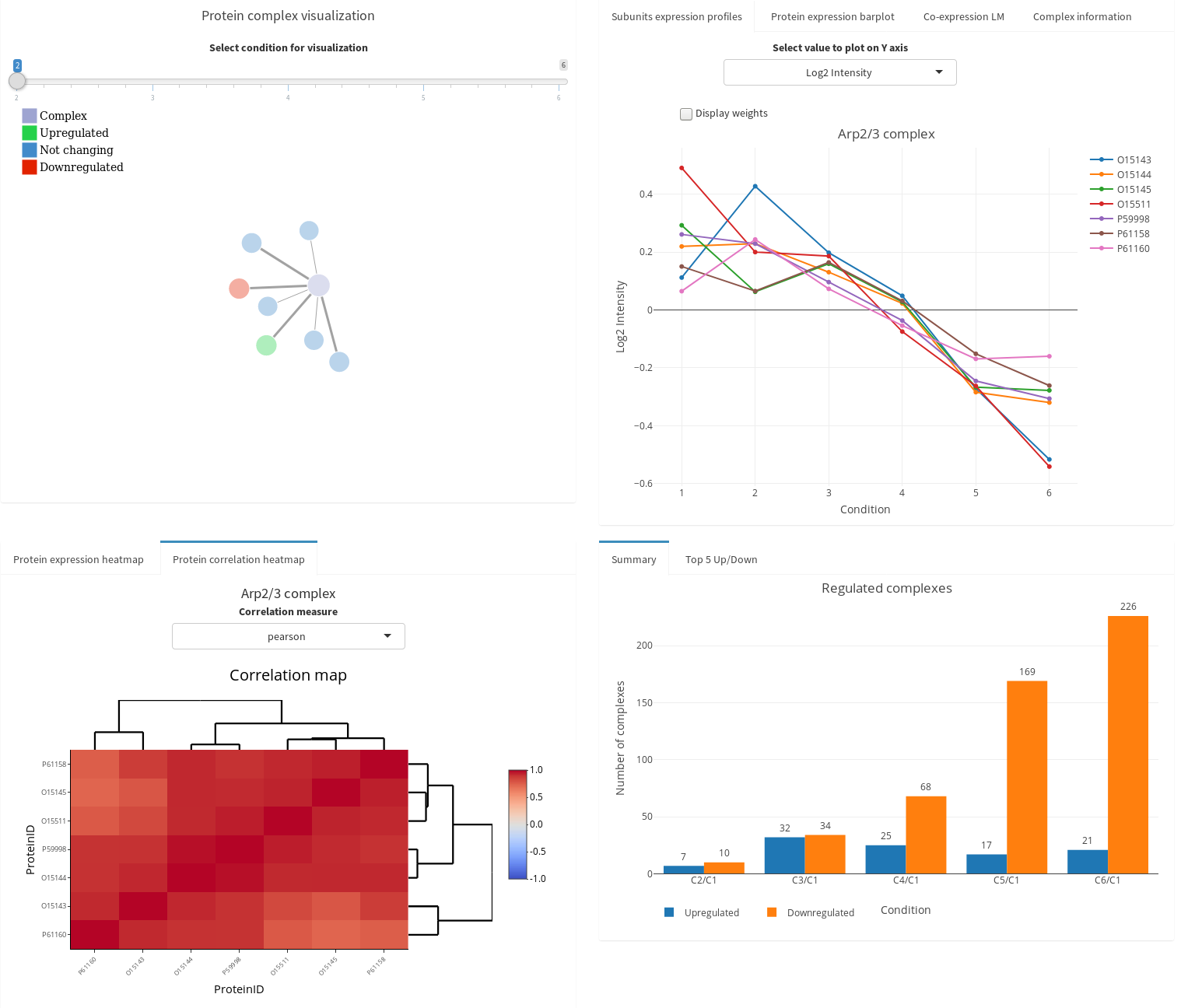
Protein Complexes
ComplexBrowser to investigate the
behavior of protein complexes in your
proteomics data set (ref).
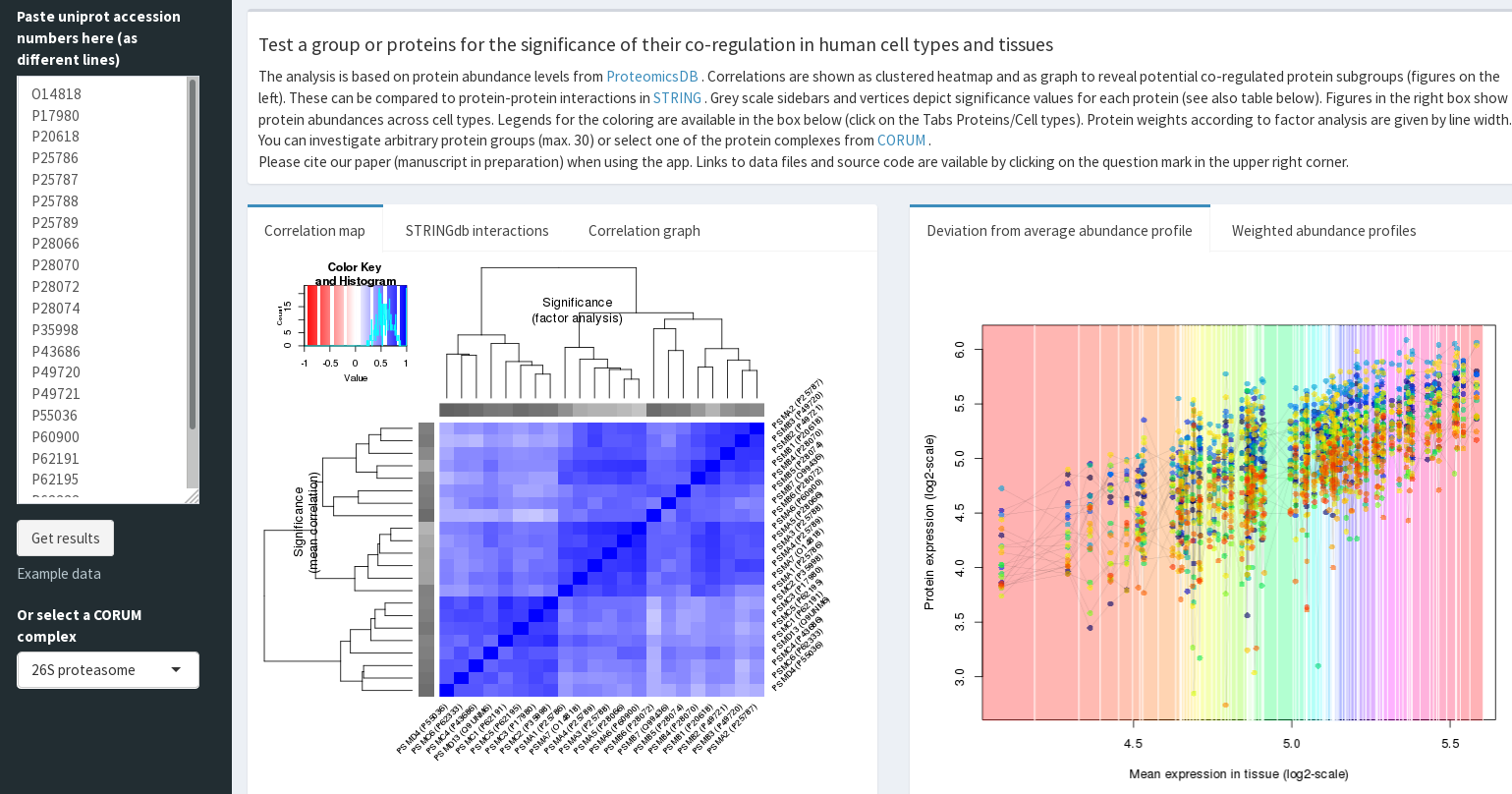
CoExpresso to look for co-regulatory
patterns within hundreds of human cell types
(ref).
PTM crosstalk
Large-scale estimation of crosstalk
between nearby residues (ref.
and ref.).
Interplay scores estimate the quantitative
crosstalk between PTMs on a protein (ref.
and ref.).
Crosstalk patterns can be
visualized by CrosstalkMapper.


Internal fragment ions
Internal fragments ions are considered crucial for the
identification of proteoforms in top-down mass spectrometry but are ubiquitous and noisy. We
show
how they can be used to
validate proteoforms (ref).
Middle-down mass spectrometry
We develop and apply a workflow to quantify PTMs
on histone tails
(refs).
ProteoformQuant is under active development for improved quantification of proteoforms in
middle-down MS data.
CrosstalkDB
With quantitative data
from middle-down and top-down mass
spectrometry, the web server collects and
analyzes the input files, followed by
statistical assessment of the crosstalk between
measured PTMs
(refs).
Computational models
Taking simple
rules for writing, propagating and deleting
histone PTMs on chromatin, we were able to
reproduce global patterns measured by ChIP-seq
experiments. The implementation of crosstalk
rules results in a rich spatial and temporal
behavior (ref).

Peptide representations for deep learning
In collaboration with the
Röttger group, we developed an environment
to retrieve millions of mass spectra (MS1 and
MS2) from the public repository PRIDE, to
categorize them in a database, and to create
data representations that can be directly used
for machine learning purposes: MS2AI
(refs).
Blind application of deep learning methods to MS leads to high bias and inaccurate predictions. We investigated the impact of data variability on the prediction bias in (ref).
See also the AIMe registry to report AI-based biomedical results in a standardized and reproducible manner (ref) and our community paper about machine learning in proteomics (ref).


Standardization and community efforts
See the proposed notation of proteoforms:
ProForma (ref).
Accurate metadata annotation is crucial for ensuring reproducibility and data repurposing.
Together with the EuBIC community, we developed
a data standard for proteomics metadata: (refs.).
As part of ELIXIR DK, we annotate proteomics
tools for
bio.tools
(refs.) and are part of the proteomics
community (see also white paper, ref).
We also recently contributed to a community perspective on preventing proteomics data
tombs via shared standards, incentives, and stewardship
(ref).
WOMBAT-P
We implemented four
scalable and portable workflows for
label-free data analysis as part of an
ELIXIR implementation study. They allow to
systematically compare the performance of
different data analysis workflows (ref).
Antibodies
Antibodies are notoriously difficult to identify due to their large sequence variety. Taking
advantage of the
Observed Antibody Space, we developed
a
workflow to
identify antibodies in mass spectrometry data (ref).
ProtProtocols
As part of a project
within the EuBIC initiative, we developed a framework
for fully reproducible, documented and
user-friendly pipelines for specific cases of
proteomics data analysis. Within this
framework, we created IsoProt, a full
data analysis pipeline for iTRAQ/TMT data
(ref).
Check it out here: IsoProt
at GitHub – download it via our
docker-launcher
Biological evolution
See my former
studies of aging, sympatric speciation and
competitive cellular automata (refs).
Simulations
Almost anything can be
simulated on the computer including sand dunes,
opinion dynamics and linguistics (refs).
Statistical Mechanics
See my work on
generalized entropies and Fokker-Planck
equations (refs).
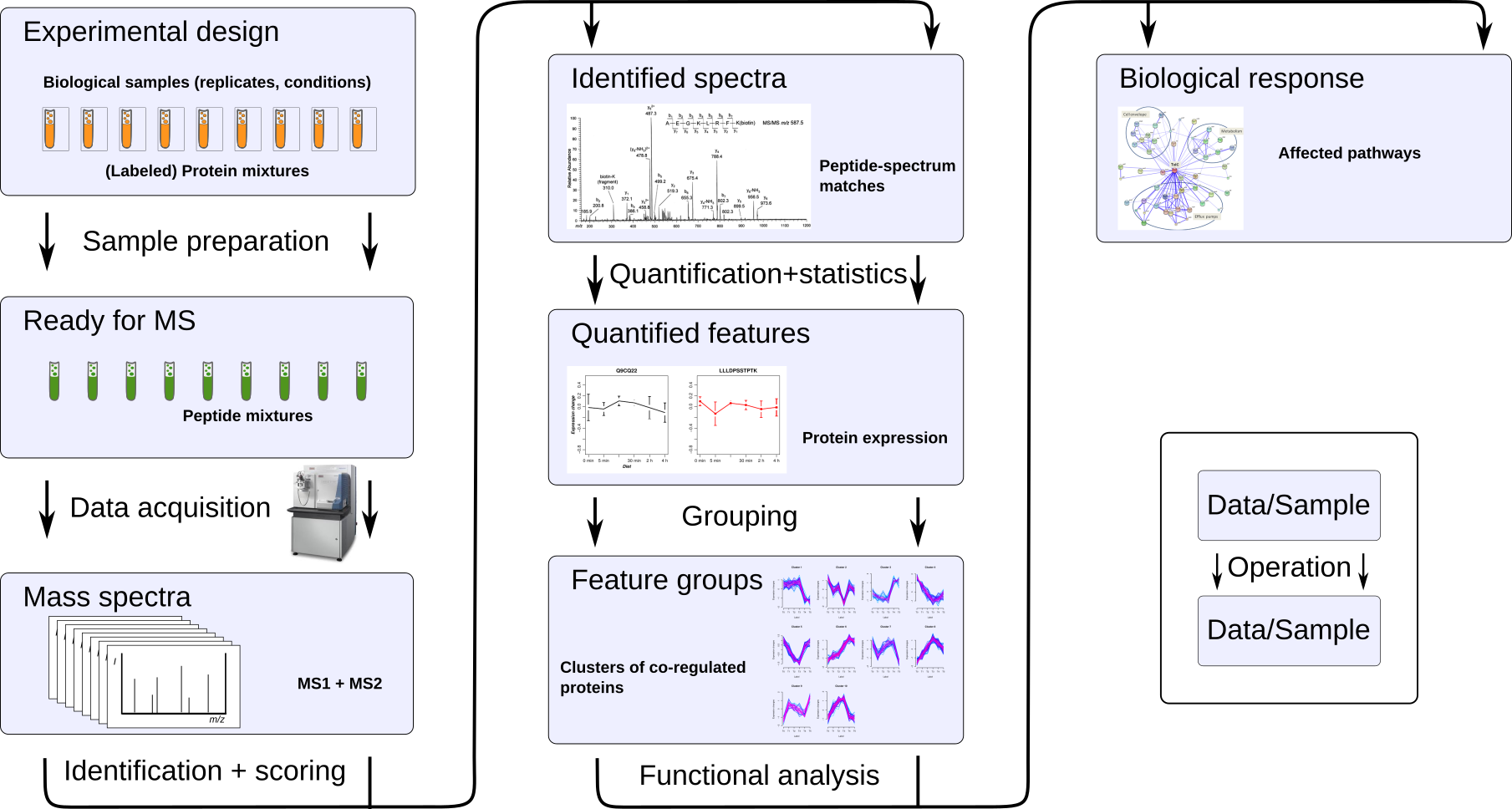
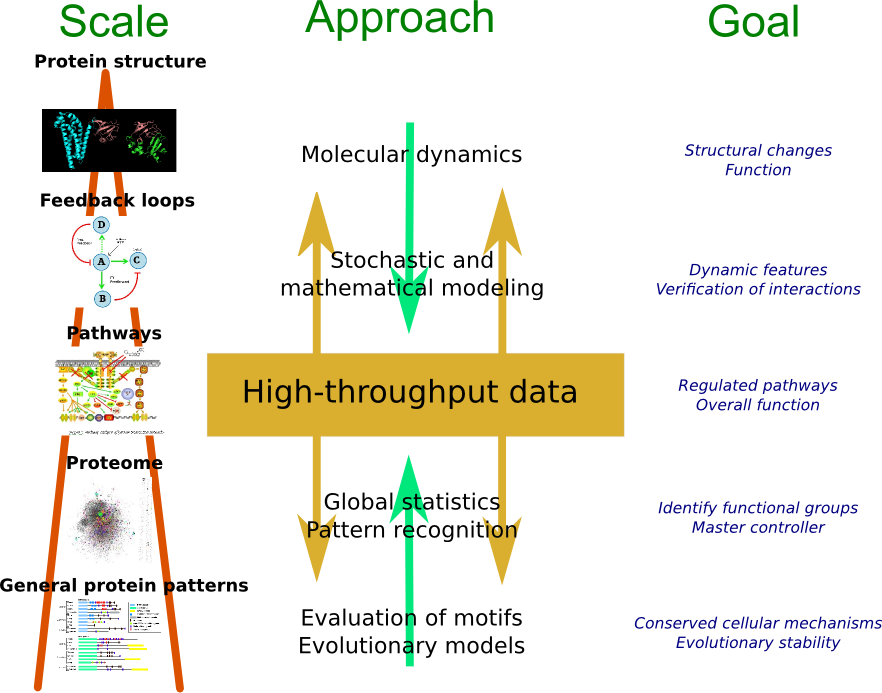
An overview of the main methods for analysis of data from peptide mass spectrometry and other -omics data.
My group runs and co-teaches multiple courses at the Department of Biochemistry and Molecular Biology and beyond:
We offer projects for Bachelor and Master students. To get an idea, take a look at our research topics. Interested? Contact us.
First year bachelor projects
Bachelor projects
Master projects
All applications are hosted on this server and accessible directly from your browser. If an app is
temporarily unavailable due to high load, please try again later or run it locally. Local options
include Docker containers (image names follow the pattern
veitveit/<app_name>), access via the UCloud / SDU
Cloud platform, and R packages (available for PolySTest and VSClust).
Automated pre-processing workflow with handshakes to other tools for quantitative omics datasets.
Variance-sensitive clustering of quantitative data with integrated statistical testing and pathway analysis.
Combined statistical testing for multi-omics data with few replicates and missing values.

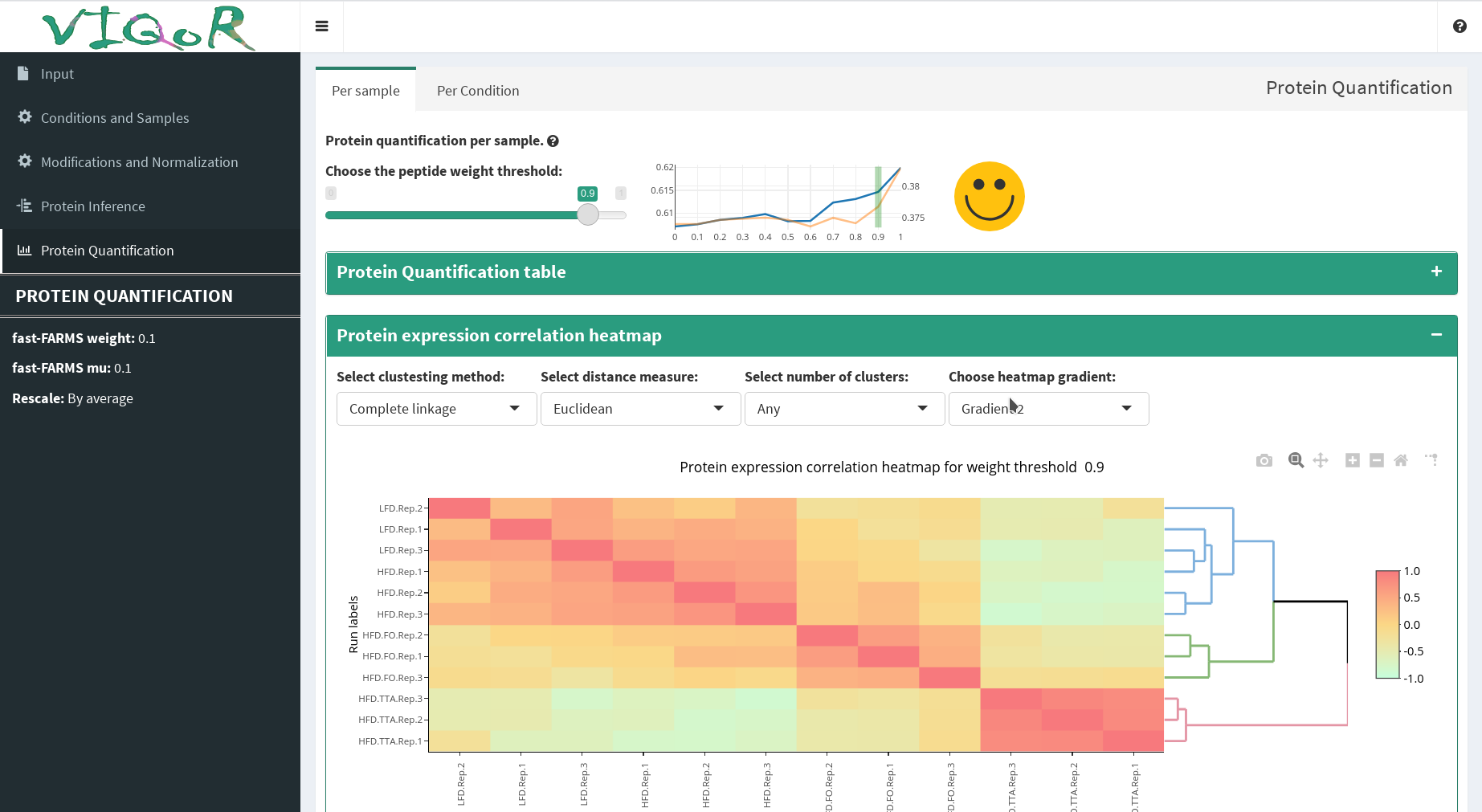
Interactive protein inference, summarization and visualization using parsimonious inference and factor-analysis-based summarization.
ELIXIR is the European Life Sciences infrastructure responsible for coordinating bioinformatics resources across Europe. ELIXIR Denmark runs bio.tools, the registry of bioinformatics tools and data resources, used by our group for software metadata annotation and discoverability.
Initiative of bioinformaticians in Europe to improve support and coordination of training and
software development in proteomics and metabolomics informatics.
Conference: We are organizing conferences, hackathons and workshops in Computational
Proteomics.
EuPA heads the national proteomics societies and organizes the EuPA conferences as well as multiple events like Summer and Winter Schools.
BalticSeaBioMed is a collaborative network in biomedicine connecting six universities around the Baltic Sea.
Activities: Joint training, and summer school activities in biomedicine and global health.
Publication list loaded automatically from EuropePMC. For the full record see also Google Scholar.
Loading publications…